What is the biologics license application or BLA?
A Biologics License Application (BLA) is submitted to the Food and Drug Administration (FDA) to get permission to distribute a biologic drug or product in the US. A BLA can be submitted by an applicant responsible for the efficacy and safety of the biological product.
A BLA is regulated under 21 CFR 600-680 and generally applies to vaccines and other allergenic drug products, blood products, including cellular and genetic therapies.
The Center for Biologics Evaluation and Research (CBER) regulates products under several regulatory authorities, including the Food Drug and Cosmetic Act and the Public Health Service Act. It also includes the BLA process, a request to permit introducing a biologic product into interstate commerce.
What is the process of BLA submission?
For BLA submission, the applicants must submit Form FDA 356h to CBER (Centre for Biologics Evaluation and Research). CBER is accountable for the regulation of biologics.
Form FDA 356h is an application required to be submitted to place biologics, new drug, or antibiotics in the market. CBER accepts both papers and e-submissions.
A BLA is submitted by any legal entity or person involved in the manufacturing of the investigational biologics or an applicant for a license who is ready to take the responsibility for compliance with the establishment and product standards.
The following information is required for BLA submission with Form 356h:
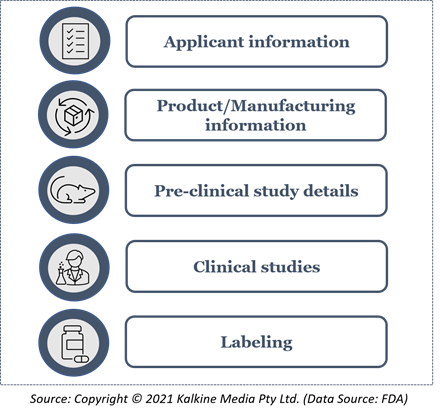
After the application submission to the FDA, the regulatory authority evaluates whether the application is complete or not. The FDA also conducts an initial review of the standard operating procedures (SOP) and the data submitted.
If required, the FDA can raise a request to the biologic product’s manufacturer for some additional information regarding any aspect.
What is the electronic submission of BLA?
The electronic submission of BLA involves the submission of the application in electronic format to the Center for Biologics Evaluation and Research. The CBER will update and modify the guidance documents on electronic submissions regularly to indicate the evolving technology. For every electronic submission, applicants need to submit a comprehensive table of contents comprising three or four levels of detail, with the proper bookmarks and hypertext links.
What are the differences between BLAs and NDAs?
While the purpose of BLAs and NDAs is the same - to gain approval for marketing a product in the US, there is a minor difference in their application content and submission requirements.
Regarding approval criteria, NDAs must fulfil the below-mentioned criteria:

Source: Copyright © 2021 Kalkine Media Pty Ltd (Data source: FDA)
Similarly, contents for a BLA should establish the safety and potency of a biological drug. BLA content must demonstrate purity, particularly to imply that the final product does not contain any extraneous material.
However, in the case of BLA, once the application is approved, the Sponsor is granted a license for the biological drug; this licensing process is not a part of the NDA.
A new drug application (NDA) is used for drugs subject to the FDC Act’s drug approval requirements, while a BLA is required for the licensing of biological products subject to licensure under the PHS Act.
Because of the manufacturing and characterising complexity of a biologic drug, the PHS Act highlights the importance of appropriate manufacturing control. The Act offers a system for all aspects of manufacturing.
What are the similarities between BLA and NDA?
- Similar to an NDA, a BLA is also submitted to the FDA to market a new biological product in the US.
- The goal of obtaining marketing approval is the same for NDAs and BLAs. Hence, they must contain enough information to demonstrate the efficacy and safety of the drug and demonstrate an ideal risk-benefit ratio for marketing approval.
- Several regulations are common and apply to both NDAs and BLAs, such as accelerated approval pathways, labeling and advertising rules, PDUFA fees and pediatric study requirements.
- Like an NDA, a BLA includes relevant findings from the clinical trials and CTD documentation, including the REMS (Risk Evaluation and Mitigation Strategies) document.
- FDA’s form 356h is applied for the submissions of both an NDA and a BLA.
NDAs and BLAs are the two types of applications submitted to market a new medicinal product in the US. While both are submitted to obtain FDA’s drug approval, they differ in terms of approval criteria, specific regulations, and product categories.
What is Expanded Access to Experimental Biologics?
Clinical trials for any drug or biologics are developmental studies conducted to assess their safety and efficacy. Patients enrol in clinical trials to gain access to investigational therapies and support in the evaluation of investigational therapy.
Getting a drug or biologics with the expanded access program may be an option for patients who cannot enrol in clinical trials. Since the 1970s, the FDA has allowed expanded access to experimental drugs and biological products.
Expanded access can provide seriously ill patients with access to investigational therapy when they do not have any other option. This access has permitted several patients having cancer, HIV/AIDS, and other indications to get promising treatments when there is no approved alternative therapy.
Notably, if there are emergency conditions requiring the patient to be treated before submission of a written application, the regulatory agency can allow the treatment to continue without a written submission, provided all the criteria for the application are met.
 Please wait processing your request...
Please wait processing your request...