What are Generics or Generic Drugs?
Generic drugs are copies of branded or innovative drugs that were developed by other healthcare companies. A generic drug is designed to be similar to already marketed branded medicines in dosage, safety, strength, route of administration, quality, as well as intended use. These versions are pharmaceutically equivalent to the brand drugs, also termed as patented drugs.
The similarities between a generic and a branded drug aid in proving the bioequivalence. Being bioequivalent indicates that a generic drug works in a similar manner & provides an identical clinical advantage as its branded counterpart.
In other words, a generic medicine acts as a substitute for the branded version. The active ingredients in generics are the same as that of the brand-name drug or an innovator drug.
Like an innovator drug, generic drugs also require regulatory approval before they can be commercialized in the market. Generic drugs also require approval from the Food and Drug Administration (FDA) before they are available for sale in the US.
Although generic drugs might not be associated with any single entity, these drugs are generally subject to government regulations in the nations where they are dispensed.
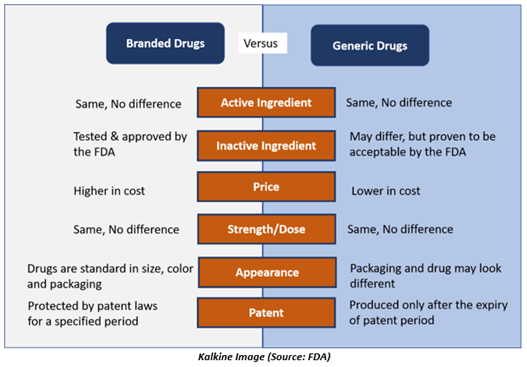
How are Generic Drugs Approved?
For a new or innovative drug, the sponsor/company seeking an approval needs to file a New Drug Application (NDA). However, for generic drugs, the application submitted to the FDA is known as an Abbreviated New Drug Application, or ANDA.
Similar to any other scientific and regulatory process, generic drug approval takes time.
The FDA requires time to review the complex information necessary to prove that a given generic drug can be substituted for the patented or brand drug that it copies. The time also depends on drug product complexity and the completeness of the application.
The application submitted to the FDA seeking approval should demonstrate that:
- The generic drug is pharmaceutically equivalent to the patented drug.
- The manufacturer is proficient at manufacturing the drug accurately and consistently.
- The active pharmaceutical ingredient (API) is the same as that of the brand.
- The right amount of the API gets to the place in the body where it has an effect.
- The inactive ingredients present in the drug are safe.
- The container of the drug in which it will be shipped & marketed is appropriate.
- The label of the generic drug is the same as the brand-name drug’s label.
- Relevant patents or legal exclusivities related to brand-drug are expired.
The Generic Drugs Program of the FDA performs a comprehensive review to make sure generic medicines meet all the requirements. The generic medicines that are approved by the regulatory agency are generally marketed after patents and exclusivities of the branded drug expire.
What is an ANDA?
An Abbreviated New Drug Application, or ANDA, is submitted to the Food and Drug Administration for the review & approval of generic drugs. FDA’s Office of Generic Drugs regulates abbreviated New Drug Applications. The applications are considered abbreviated, as for ANDA, there is no requirement to include information from preclinical and clinical trials to determine safety and efficacy of the drug.
For an ANDA, the applicants must scientifically demonstrate that their drug performs in a similar way as the innovator drug product. Once approved, the applicant can produce and market the generic drug to provide a safe, effective alternative to a branded drug it mimics at a significantly lower cost.
Companies manufacturing generics must provide supporting documents demonstrating that the active ingredient in generic drugs is similar to that of the brand drug, and FDA must review that proof.
An ANDA must demonstrate the generic medicine is equivalent to the brand drug in following ways-

Why do Generics cost significantly less than the Branded Drugs?
Generic drugs become available only after a thorough review by the FDA and after a set period that the brand drug has been on the market exclusively. This is because new drugs are generally protected by patents that prohibit others from making as well as selling copies of the patented drug.
Generic drugs cost less than their branded peers because the applicants do not have to perform any animal study and clinical trials that were necessary for the brand-name drug to prove safety & effectiveness.
Are the side effects of generic drugs monitored?
After the FDA approves any innovative or generic medicine, the agency continues to investigate the safety of the medicine. FDA takes several actions to ensure safety as well as quality before and after the marketing of a new medicine or generics.
The staff of the agency examines drug products to make sure the medicines are safe and effective with high quality at all levels of the supply chain, from APIs to final products being marketed to consumers.
Any side effects are found during the investigations may lead to a change in the manufacturing process of the product (brand-name or generic counterparts). Because of restricted resources, the Food and Drug Administration is not able to operate separate clinical trials for generic drugs.
 Please wait processing your request...
Please wait processing your request...